Með reglugerðinni er ábyrgð rekstraraðila aukin, þ.m.t.framleiðenda og dreifingaraðila, betri yfirsýn skráningaryfirvalda er tryggð, markaðseftirlit aukið, ásamt því að rekjanleiki tækjanna allt frá framleiðslu til dreifingaraðila er tryggður.
Reglugerð um lækningatæki til sjúkdómsgreiningar í glasi (ESB) 2017/746 er lögfest með lögum um lækningtæki nr. 132/2020.
Helstu breytingar með nýrri reglugerð
- Ríkari skyldur eru gerðar til rekstraraðila (framleiðenda, innflytjenda og dreifingaraðila), þ.e. varðandi rekjanleika, skráningu tækja og sannprófun á réttum merkingum.
- Flokkunarkerfi tækja breytist í takt við tækniframfarir og til þess að auka öryggi sjúklinga. Flokkun tækja mun ákvarða viðeigandi samræmismatskröfur. Tæki munu flokkast í A, B, C eða D flokk eftir áhættu, þar sem tæki í flokki A eru áhættuminnsti flokkurinn og tæki í flokki D eru áhættumesti flokkurinn. Rétt flokkun tækja er á ábyrgð framleiðanda.
- Aukið eftirlit er með lækningatækjum til sjúkdómsgreiningar í glasi áður en þau eru sett á markað. Lækningatæki til sjúkdómsgreiningar í glasi eru nú háð eftirliti tilkynntra aðila sem þurfa að votta tæki m.t.t. nýrrar flokkunar. Tæki í flokki A eru undanskilin kröfu um tilkynnta aðila nema þau séu dauðhreinsuð. Vottorð skulu gefin út af tilkynntum aðila.
- Gerð er krafa um að fleiri skilyrði séu uppfyllt vegna tilnefningar á tilkynntum aðilum og aukið eftirlit með þeim.
- Gefa þarf út nýjar samræmisyfirlýsingar (EU Declaration of conformity) fyrir tæki í samræmi við kröfur nýrrar reglugerðar sem eiga að innihalda viðbótarupplýsingar eins og einkvæmt tækjaauðkennisnúmer og þar sem við á tilvísanir í sameiginlegar forskriftir.
- Bætt gagnsæi með víðtækum gagnagrunni ESB um lækningatæki (EUDAMED), en hlutar hans verða aðgengilegir almenningi
- Rekjanleikakerfi byggt á einstöku tækjaauðkenni (UDI);
- Auknar kröfur vegna virkni prófana til að sannreyna greiningarvirkni eða klíníska virkni tækis.
- Auknar kröfur til framleiðenda um að sinna eftirliti með þeim lækningatækjum sem hann framleiðir eftir markaðssetningu
- Sérstakt fyrirkomulag fyrir tæki sem eru framleidd og notuð á sömu heilbrigðisstofnun („in-house devices“).
Rekstraraðilar kynni sér lykildagsetningar
Það er mikilvægt fyrir rekstraraðila sem koma að samræmingu tækja við lög og reglugerðir að kynna sér lykildagsetningar varðandi vottanir tækja, þ.e. hvenær tæki þurfa að vera komin með nýja samræmisyfirlýsingu samkvæmt reglugerðinni.
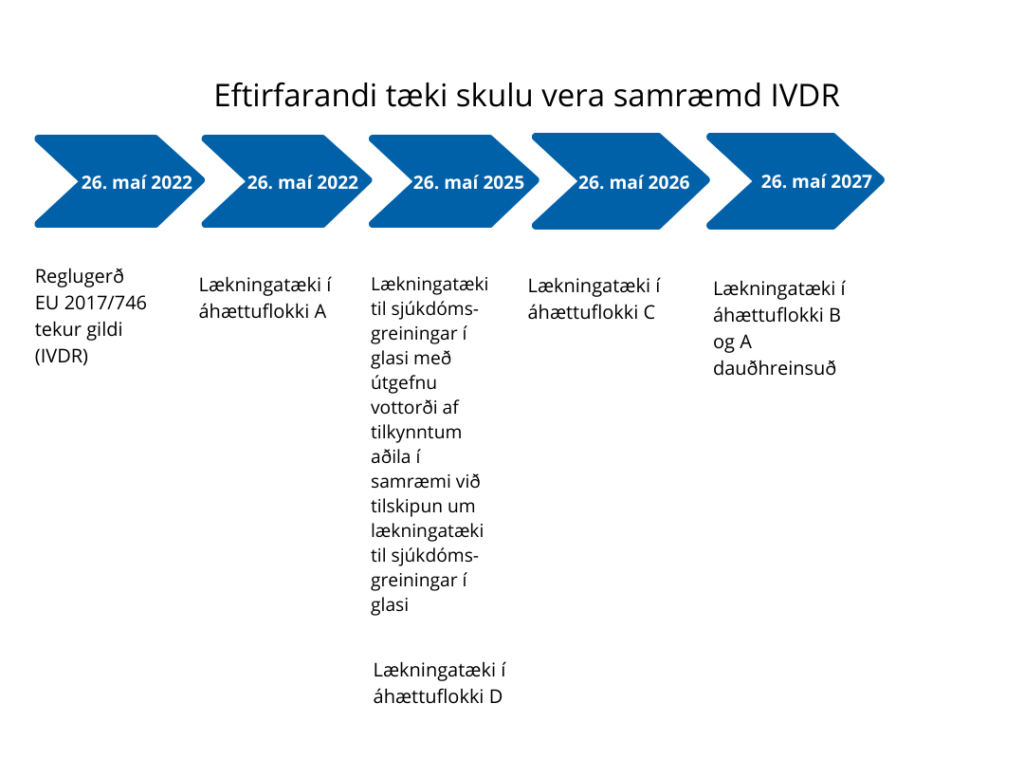
Aðlögunartímabil lækningtækja til sjúkdómsgreiningar í glasi sem vottuð hafa verið af tilkynntum aðila ásamt tækjum sem þurfa að gangast undir samræmismat tilkynntra aðila í fyrsta skipti er til 26. maí 2025. Fer aðlögunartímabil eftir áhættuflokki tækis:
Mikilvægt að tilkynna atvik til Lyfjastofnunar
Lyfjastofnun ítrekar að öllum þeim sem framleiða, selja, dreifa, eiga eða nota lækningatæki og vita um atvik, frávik, galla eða óvirkni, sem kynni að valda eða hefur valdið heilsutjóni eða dauða notanda, ber skylda til að tilkynna Lyfjastofnun um slíkt.
Spurningar og svör um reglugerð um lækningatæki til sjúkdómsgreiningar í glasi (IVDR)
Reglur Evrópusambandsins (ESB) um markaðsetningu lækningatækja, þar með talin lækningatæki til sjúkdómsgreiningar í glasi, voru settar á tíunda áratug síðustu aldar. ESB lét uppfæra reglurnar til að mæta tæknilegum og vísindalegum framförum á þessu sviði síðustu 20 árin með það að markmiði að reglurnar væru í takt við tímann og auka öryggi lækningatækja.
IVDR tók gildi þann 26. maí 2022 og með reglugerðinni er leitast við að tryggja lýðheilsu og öryggi sjúklinga m.t.t. framfara í vísindum, auk samræmdrar starfsemi innan lækningatækjamarkaðsins. Reglugerðin gekk í gildi samhliða reglugerð um lækningatæki (MDR) 26. maí 2021.
Lækningatæki til sjúkdómsgreiningar í glasi eru notuð til prófunar á lífsýni til að ákvarða heilsufar einstaklings. Mikið úrval er af greiningarprófum (IVDs), allt frá þungunaprófum og blóðprófum fyrir sykursjúka, til háþróaðra greiningarprófa sem unnið er með á klínískum rannsóknarstofum. Dæmi um það síðarnefnda eru HIV eða COVID-19 greiningarpróf.
Í 110. grein IVDR reglugerðarinnar er kveðið á um aðlögunartímabil til 26. maí 2024 fyrir þau lækningatæki til sjúkdómsgreiningar í glasi sem hafa vottorð frá tilkynntum aðila samkvæmt eldri tilskipun. Þetta aðlögunartímabil er framlengt um 1 ár, eða fram í maí 2025. Framlengingin mun því aðeins gilda fyrir tæki sem hafa vottun samkvæmt eldri tilskipun, eða 8%“
Ástæða fyrir lengingu aðlögunartímabilsins, er annars vegar álags sem hefur skapast vegna COVID-19 heimsfaraldursins og hins vegar þær umfangsmiklu breytingar sem fylgja IVDR reglugerðinni. Þetta leiddi til að yfirvöld aðildarríkja ESB, heilbrigðisstofnanir, tilkynntir aðilar og rekstraraðilar höfðu ekki uppfyllt að fullu skilyrði IVDR reglugerðarinnar við gildistöku hennar 26. maí 2022.
Jafnframt leggur framkvæmdastjórn ESB til lengra aðlögunartímabil fyrir tæki sem þurfa að gangast undir samræmismat þar sem tilkynntir aðilar taka þátt í fyrsta skipti samkvæmt IVDR reglugerðinni. Í þeirri tillögu er gerður greinarmunur á áhættuflokkum. Aðlögunartími fyrir tæki sem teljast vera með mikla áhættu (flokkur D) verður fram í maí 2025. Aðlögunartími fyrir tæki í flokki C verður fram í maí 2026, og aðlögunartími fyrir tæki með minni áhættu (flokkur B og A dauðhreinsuð) verður fram í maí 2027. Þessi leið miðar að því að feta einstigið milli aðkomu tilkynnta aðila og lýðheilsusjónarmiða.
Nei, almennur innleiðingardagur IVDR reglugerðarinnar var 26. maí 2022.
Frá og með þeim tíma gildir reglugerðin um lækningatæki til sjúkdómsgreiningar í glasi sem þegar eru CE-merkt og krefjast ekki aðkomu tilkynnts aðila, þ.e. tæki í flokki A og ný tæki sem sett eru á markað það eru þau tæki sem eru ekki með vottorð frá tilkynntum aðila né samræmisyfirlýsingu frá framleiðanda sem gefin er út fyrir 26. maí 2022.
Tæki þar sem lengra aðlögunartímabil á við um, má aðeins nota að því tilskildu að þau uppfylli ákveðin skilyrð um að þau uppfylli áfram skilyrði tilskipunina og að engar verulegar breytingar séu á hönnun þeirra og fyrirhuguðum tilgangi.
Ennfremur, frá innleiðingadegi reglugerðarinnar, munu hertar reglur um gátartilkynningar og markaðseftirlit einnig gilda um tæki sem falla undir skilgreiningu aðlögunartímabils.
Nei, það er ekki tilgangurinn með aðlögunartímabilinu. Það er mikilvæt að framlengja aðlögunartímabilið til að ekki dragist úr framboði á nauðsynlegum lækningatækjum til sjúkdómsgreiningar í glasi, þannig að aðgengi sjúklinga að viðeigandi heilbrigðisþjónustu sé ekki í hættu.
Aðlögunartímabilið á aðeins við um þær vörur sem eru á markaði samkvæmt núgildandi tilskipun 98/79/EB um lækningatæki til sjúkdómsgreiningar í glasi, þ.e. fyrir 26. maí 2022 og eru því taldar öruggar og með trygga verkun. Ný lækningatæki til sjúkdómsgreiningar í glasi, og tæki þar sem hönnun eða ætlaður tilgangur hafa breyst þurfa að vera í samræmi við kröfur IVDR.
Enn fremur verða framleiðendur að búa sig undir vottunarferli samkvæmt IVDR eins fljótt og auðið er. Þeir skulu aðlaga gæðastjórnunarkerfi, vörur, tæknigögn og tryggja aðkomu tilkynnts aðila löngu fyrir lok aðlögunartímabilsins.
Ef framleiðandi CE merkts lækningatækis missir CE merkingu er hægt að sækja um undanþágu fyrir notkun þess skv. 54. gr. IVDR reglugerðar. Frekari upplýsingar um umsóknarferlið, hvaða kröfur þarf að uppfylla og kostnað vegna hennar má finna á vef Lyfjastofnunar.
Ef heilbrigðisstofnun ákveður að framleiða lækningatæki til sjúkdómsgreiningar í glasi þarf að uppfylla allar þær kröfur sem settar eru fram í 5.lið 5. gr. IVDR.
Það er leyfilegt að uppfylltum ákveðnum skilyrðum. Það er á ábyrgð þeirrar heilbrigðisstofnunnar sem framleiðir slík tæki að uppfylla kröfur sem settar eru fram í 5. lið 5. gr. IVDR. Samkvæmt þeim geta aðildarríkin krafist þess að heilbrigðisstofnanir leggi fyrir viðkomandi yfirvald allar frekari upplýsingar sem máli skipta um þau tæki sem hafa verið framleidd og notuð á þeirra vegum.
Aðildarríkin skulu einnig halda þeim rétti að takmarka framleiðslu og notkun á tiltekinni gerð slíkra tækja, og þeim skal veittur aðgangur til að skoða starfsemi viðkomandi heilbrigðisstofnana.
Lækningatæki til sjúkdómsgreiningar í glasi sem eru framleidd á heilbrigðisstofnun þurfa ekki vottun tilkynnts aðila. Heilbrigðisstofnunin skal uppfylla öll atriði sem talin eru upp í 5.lið 5. gr. IVDR.
Sönnunarbyrðin fellur á þá heilbrigðisstofnun sem framleiðir lækningatæki til sjúkdómsgreiningar í glasi. Skv. D lið 5. liðs 5. gr. IVDR skal heilbrigðisstofnunin rökstyðja í gögnum sínum að ekki sé hægt að uppfylla sérstakar þarfir tiltekins markhóps sjúklinga, eða að ekki sé unnt að uppfylla þær á fullnægjandi hátt með sambærilegu tæki sem fáanlegt er á markaði.
Lyfjastofnun tekur ekki sama lista yfir lækningatæki, en með tilkomu EUDAMED verður m.a. haldið miðlægt utan um skráð tæki frá framleiðendum. Rekstraraðilar sem annast umsýslu lækningatækja, s.s. dreifingaraðilar og innflytjendur, gegna ákveðnum skyldum skv. 13. og 14. gr. í IVDR. Skylda rekstraraðila er m.a. að gæta þess að tækið sé CE merkt og að útbúin hafi verið ESB samræmisyfirlýsing fyrir tækið.